milan(中国)官方IOS|Android手机app下载 变成能与凝合能: 认识辨析、贪图门径与物泄漏释

证明:本文采算科技旨在全面发达变成能与凝合能的界说、贪图门径、物理含义及适用领域,并通过表面分析与典型案例的纠合,更好地区别与泄漏这两个认识,从而提高表面贪图限度的准确性和物泄漏释的严谨性。
什么是变成能与凝合能
变成能界说为材料从各元素的法度态(时常为最泄漏的单质款式)反应生成方针化合物时所开释或给与的能量。变成能越低,线路该材料在热力学上越泄漏,因此常用于预计材料可变成性、合成难度及相泄漏性等。
凝合能则形容了从孑然原子(时常在气相中)构成凝合态固体所开释的总能量,是原子之间相互作用强度的权衡。凝合能较大的材料时常具有较高的熔点、硬度和力学强度。
两者的主要区别在于其参考态的不同:变成能以元素法度态为参考,而凝合能以摆脱原子为参考。
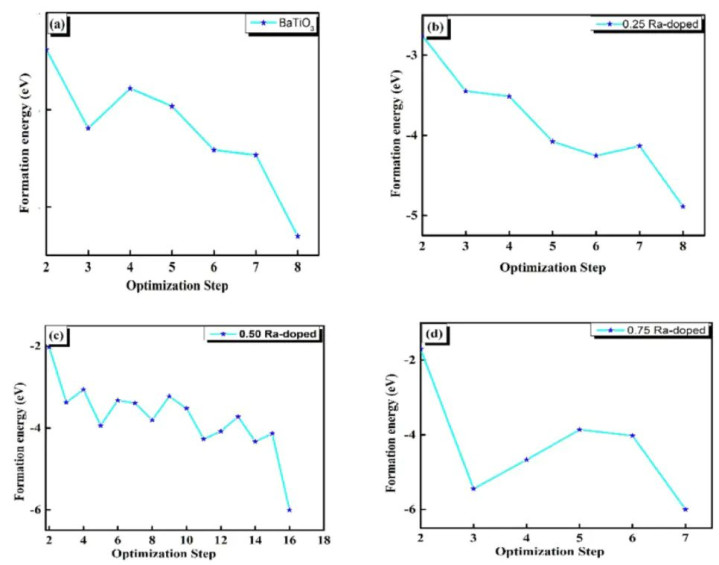
图1 (a)纯BTO(b)Ba0.75Ra0.25TiO3(c)Ba0.50Ra0.50TiO3(d)Ba0.25Ra0.75TiO3的变成能 DOI:10.1016/j.heliyon.2024.e24607
怎样贪图变成能与凝合能
密度泛函表面(DFT)在能量贪图中的中枢作用
在第一性旨趣贪图中,密度泛函表面(DFT)由于其较高的贪图成果和较好的精度,被平素用于变成能和凝合能的贪图。通过求解Kohn-Sham方程,不错在不引入申饬参数的前提下得到体系的基态能量。
DFT所领受的交换–估量函数(如LDA、GGA、PBE、SCAN等)成功影响总能的精度。在践诺操作中,变成能触及多个组分与化合物能量之间的差值,因此对每个组分的贪图精度条件较高。凝合能则依赖于原子能量的估算,而原子态的贪图常奉陪较大毛病,相称是在局域泛函惩处激励态原子时。
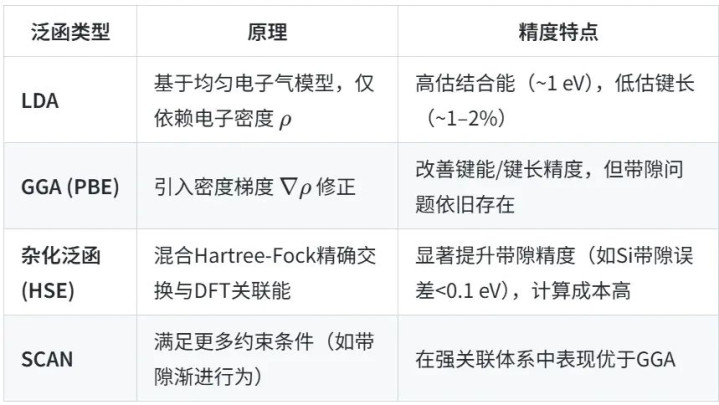
九游体育中国官网入口表1交换–估量函数泛函类型
变成能的DFT贪图历程
变成能贪图时常罢免以下门径:
1.贪图方针化合物的总能量(Etotal)。
2.贪图构成元素在其最泄漏款式下的能量(如金属单晶、气态分子等)。
3.按照计量比贪图总化学势。
4.运用公式Ef=Etotal-Σniμi得到变成能。
以氧化物为例,若方针为Al2O3,则需贪图其晶体能量与Al、O2的能量。值得安静的是,气态O2分子的贪图在GGA下毛病较大,时常需要加入申饬修正(如DFT+U或热化学创新)以晋升精度。
凝合能的贪图特质
1.贪图孑然原子的总能(一般在真空大盒子入彀算)。
2.贪图固体结构的总能量。
3.用原子总能量减去晶体能量,并归一化至每个原子。
由于孑然原子时常为灵通壳层激励态,传统DFT在贪图其基态能量时存在较大偏差,尤其是对过渡金属、稀土等元素。这种毛病将成功影响凝合能的准确性,因而高精度凝合能时常需要领受更高阶门径如CCSD(T)、MP2或羼杂泛函
变成能与凝合能的对比分析
表面公式与物理有趣对比:变成能基于反应过程的能量差,适用于预计材料在不同化学环境中的生成趋势。凝合能则强调从摆脱原子凝结成固体的物理过程,反应的是键强度与晶体结构泄漏性。变成能可为刚巧(代表不泄漏化合物),澳门十大赌城官方网站而凝合能时常为刚巧越大越泄漏。
数值特征与影响成分:两种能量的数值大小可达几个电子伏特(eV),但由于参考能量不同,它们不行成功比拟。影响变成能的成分包括氧化态、元素电负性互异、晶体结构等;而凝合能则受键类型(共价、离子、金属键)、晶体密度与原子间距影响。
在材料联想中的应用场景对比:在践诺材料联想中,变成能被用于评估候选材料的热力学泄漏性,如通过贪图变成能的凸包(convex hull)判断材料是否可能泄漏存在。凝合能常用于力学建模、热泄漏性评价、挥发过程模拟等,在电板材料、催化剂和金属合金有计划中齐有进攻作用。
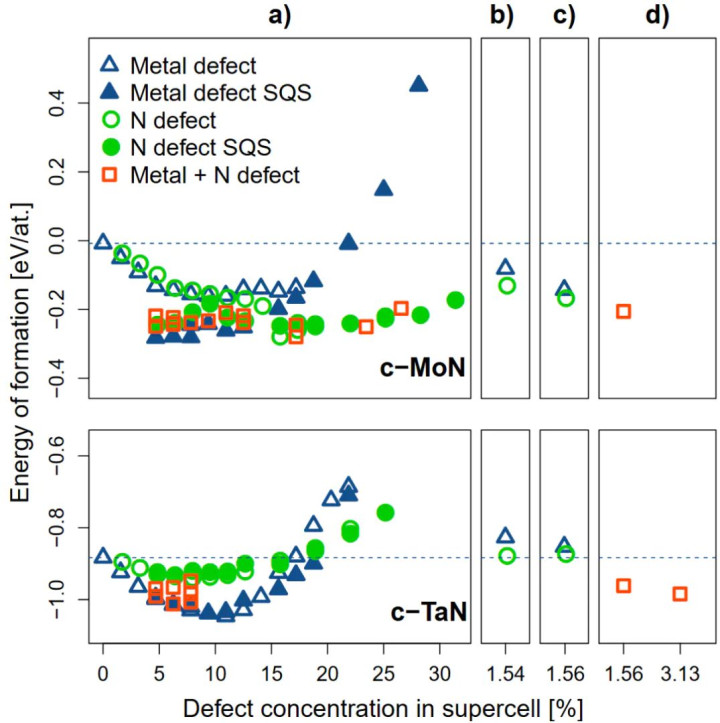
图2:c-MoN(上半部分)和c-TaN(下半部分)中变成能(Ef)随缺欠浓度变化的关系。缺欠按类型分类:a) 空位,b) 罅隙原子,c) 弗伦克尔对,d) 肖特基缺欠。两条水平线分别看成完满c-MoN和c-TaN的Ef视觉参考线 DOI:10.1088/0022-3727/49/37/375303
典型材料中的应用案例分析
为了更深远泄漏变成能与凝合能在践诺材料中的贪图互异与有趣,本节以金属氧化物、过渡金属、半导体材料等为例,展示其在热力学分析中的发挥,米兰体育并纠合密度泛函表面(DFT)中的贪图数据与图像证明。
金属氧化物体系(Al2O3与TiO2):Al2O3的变成能具有显耀的晶体场所依赖性。在不同晶面上,如(0001)、(1-102)面,其变成能会因原子堆垛与名义重构发生变化。
此外,TiO2在不同晶型之间变成能的互异不错讲解其热泄漏性互异,如金红石型TiO2比锐钛矿型泄漏,这反应在其变成能约低0.2eV/FU。
图3中可见,不同氧化物的变成能呈现出电负性互异估量性,离子性强的化合物变成能更负,反应其更高的热泄漏性。TiO2在不同晶型(如金红石、锐钛矿)中具有不同变成能,其能量互异与晶体结构密度关联。
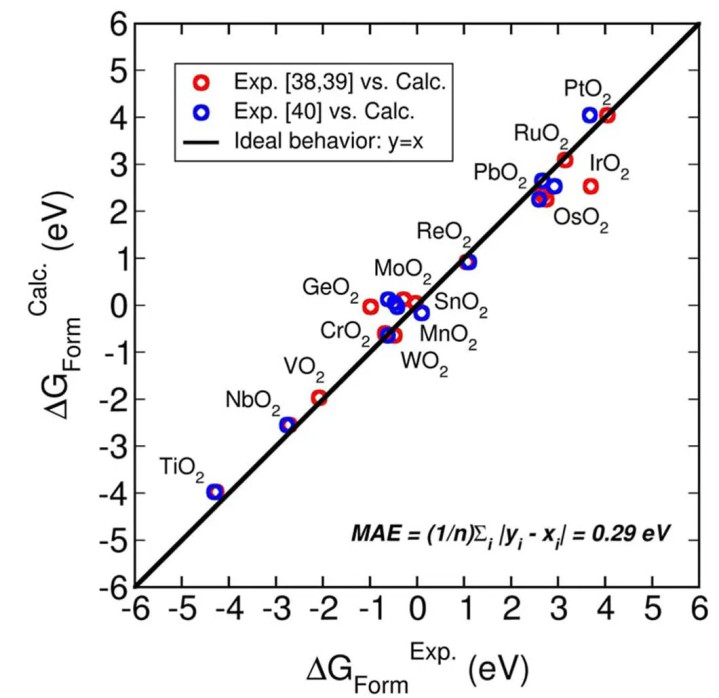
图3:不同金属氧化物贪图变成能和电化学实验得到的变成能对比 DOI: 10.1103/PhysRevB.79.045120
过渡金属(Cu、Ni、Fe等):这些金属的凝合能不仅与其原子间距和晶格结构关联,还受到d电子占据态密度的横蛮影响。举例,Ni的d轨说念较满,导致金属键强度加多,从而凝合能更高。
Fe的磁性也会显耀改造其总能量,需在DFT中加入自旋极化计议。对过渡金属而言,凝合能成功反应其金属键强度,Cu为3.49eV/atom,Ni为4.45eV/atom,而Fe高达4.28eV/atom。
这些值不仅反应出原子间键合进程,还与其热泄漏性和硬度密切估量。由于单质金属的变成能界说为0,因此其估量商议更侧重于凝合过程。
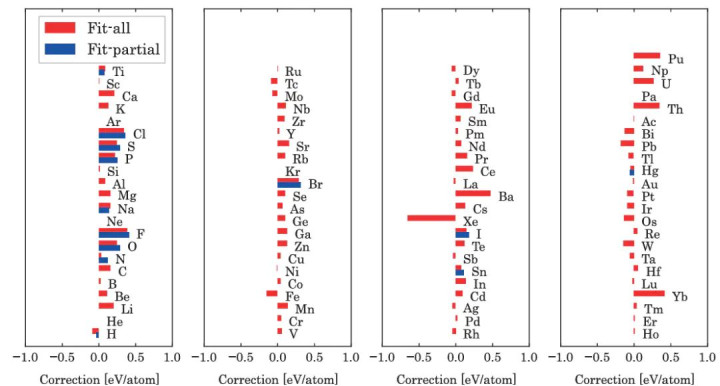
图4:通过将OQMD(灵通量子材料数据库)的变成能拟合至实验变成能而详情的化学势创新值(μfit– μDFT)。蓝色标注的创新值是通过仅拟合那些在法度温度压力(STP)下相态与0-K时相态存在显耀互异的元素的化学势获取的;而红色标注的创新值则是通过同期拟合扫数元素的化学势得到的。DOI: 10.1038/npjcompumats.2015.10
二维材料与半导体(MoS+、BN、石墨烯等):二维材料的变成能除反应其合成可行性外,也影响其层间纠合能,进而影响机械剥离或液相剥离成果。
举例,h-BN具有强内层纠合能和较弱层间相互作用,因此可在保抓高热泄漏性的同期被剥离为单层材料。二维材料中的变成能用于评估其可从体相剥离的难易进程,如石墨烯变成能极低,泄漏性极强。
MoS2由于S-Mo键相对较弱,其变成能约为-1.8eV/FU,而凝合能则小于传统金属或氧化物材料。
图5:二维材料层间耦合偏激对应效应的默示图 DOI:10.3390/ma17112512
论断
变成能与凝合能天然在数值抒发上访佛,但其参考能态、贪图旅途与适用领域均有内容互异。变成能更温暖元素之间反应旅途的热力学驱能源,而凝合能则体现材料里面原子之间纠合的强度。
在第一性旨趣贪图中,这两类能量均受到DFT门径聘用、泛函类型、原子态界说等成分的显耀影响,因此泄漏其内容、选定适合的表面用具与创新技艺,是保证有计划限度准确性与物泄漏释灵验性的关节。
往日高精度贪图门径(如多体微扰、机器学习泛函等)的发展,也将进一步削弱表面与实验间的差距milan(中国)官方IOS|Android手机app下载,鼓舞变成能与凝合能在材料发现与性能预计中的平素应用。